

BrainStorm kondigt positieve toplineresultaten aan van de VS fase 2-studie met NurOwn® bij patiënten met amyotrofische laterale sclerose (ALS)
18-07-2016
NurOwn® is aantoonbaar veilig en verdraagbaar
Klinisch betekenisvol voordeel aangetoond door hogere respons op NurOwn® vergeleken bij placebo op alle tijdpunten
Telefonische vergadering en live webcast om 10u00 oostelijke tijdzone
HACKENSACK, N.J. en PETACH TIKVAH, Israël, /PRNewswire/ -- BrainStorm Cell Therapeutics Inc., een toonaangevende ontwikkelaar van stamceltherapieën voor neurodegeneratieve ziektes bij volwassenen, kondigde vandaag de resultaten aan van de onlangs voltooide gerandomiseerde, dubbelblinde en placebogecontroleerde fase 2-studie in de VS van NurOwn® bij ALS-patiënten. De studie haalde haar primaire doelstelling. Ze toonde aan dat NurOwn veilig is en goed wordt verdragen. NurOwn haalde ook diverse secondaire efficiëntie-eindpunten. Dat bewijst duidelijk dat het product klinisch betekenisvolle voordelen biedt. Vooral van belang is dat de responsratio's hoger waren voor met NurOwn behandelde deelnemers vergeleken bij placebo op alle tijdpunten in deze studie van 24 weken.

"Deze fase 2-gegevens zijn bemoedigend. Ze tonen aan dat de NurOwn-celtherapie veilig is en dat één enkele toediening een tijdelijke en klinisch betekenisvolle respons oplevert in termen van zowel de ALS-FRS-R-meetschaal als de CSF-biomarkers", zegt dr. Robert H. Brown, Professor and Hoofd Neurologie aan de University of Massachusetts Medical Center and Medical School, en tevens onderzoeker tijdens de test. "Deze opwindende bevindingen geven duidelijk aan dat het aangewezen is een langere studie uit te voeren met repetitieve doses."
"We zijn opgetogen over de testresultaten. Ze leveren belangrijke informatie over de veiligheid, de voorafgaande efficiëntie en de biologische effecten van NurOwn", aldus Merit E. Cudkowicz, MD, MSc, Professor Neurologie aan de Harvard Medical School en Hoofd Neurologie van het Massachusetts General Hospital. "Meerdere deelnemers in de behandelde groep vertoonden een vertraagde voortgang en er waren geen veiligheidsrisico's. De verhoogde niveaus van groeifactoren in het cerebrospinaal vocht en de afgenomen ontstekingsmarkers die na twee weken werden vastgesteld, vormen bemoedigend bewijsmateriaal voor een biologisch effect. Op basis van deze resultaten zijn herhaalde doses tussen de 8 à 12 weken en een uitgebreidere bevestigende test aangewezen. Ik kijk al uit naar een voortgezette nauwe samenwerking met Brainstorm bij de ontwikkeling van NurOwn voor ALS."
"Ik voer al bijna dertig jaar klinische studies uit op het vlak van ALS en ik vind deze testresultaten persoonlijk erg bemoedigend", stelt Anthony J. Windebank M.D, Professor Neurologie and Directeur voor Ontdekkingen aan het Mayo Clinic Center for Regenerative Medicine. "We stellen positieve signalen vast op het vlak van efficiëntie en duidelijke indicaties dat het de moeite loont tot de volgende fase over te gaan. Wij en onze medewerkers van de Mayo Clinic willen dit werk maar al te graag binnen de kortste keren voortzetten."
"De patiënten die deelnamen aan de Brainstorm-studie verdroegen de behandeling bijzonder goed en de therapie had geen ernstige nadelen. Het veiligheidsprofiel biedt in ieder geval de gelegenheid om deze aanpak van de ALS-behandeling verder te bestuderen", verklaart Carlayne E. Jackson, MD, FAAN, Professor Neurologie en Otolaryngologie, Hoofd Medische Dienst van het UT Medicine San Antonio, University of Texas Health Science Center, die ook voorzitter was van het Gegevensveiligheidscontrolecomité (Data Safety Monitoring Board (DSMB)) tijdens deze studie.
"Ik zou alle patiënten die deelnamen aan de klinische test, de onderzoekers, de DSMB-leden en iedereen die betrokken was bij de succesvolle afloop van deze test willen bedanken. Deze gegevens bieden ons waardevolle inzichten in het gunstige behandelingseffect van NurOwn", stelt Chaim Lebovits, CEO van BrainStorm. "Deze studie haalde haar doelstellingen. Ze toonde aan dat NurOwn veilig is en ALS-patiënten klinische voordelen biedt. Belangrijker nog, ze zal ons helpen de studiepopulatie en het ontwerp te bepalen van een baanbrekende studie met meerdere doses NurOwn bij ALS."
Studie-ontwerp
De fase 2-test was een gerandomiseerde, dubbelblinde, placebogecontroleerde studie, uitgevoerd door meerdere centra. Ze werd ontworpen om de veiligheid en efficiëntie te evalueren van NurOwn bij 48 ALS-patiënten en werd uitgevoerd op drie locaties in de VS: het Massachusetts General Hospital, de UMass Medical School en de Mayo Clinic. De patiënten werden gerandomiseerd bij de toediening van NurOwn-cellen. Die toediening gebeurde via een gecombineerde intramusculaire en intrathecale injectie (n= 36), of placebo (n=12). De patiënten werden gedurende drie maanden maandelijks opgevolgd vóór de behandeling en zes maanden na de behandeling, waarbij ze na 2, 4, 8, 12, 16 en 24 weken werden beoordeeld. De primaire doelstelling van de studie was veiligheid en verdraagbaarheid. De vooraf bepaalde efficiëntieanalyses waren de volgende: verandering in de scorecurve van de amyotrofische-laterale-sclerose-functionele-meetschaal (Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R)), verandering in de trage vitale capaciteit (Slow Vital Capacity (SVC)) en spierkracht, responderanalyse (het percentage deelnemers dat er na de behandeling op vooruitging vergeleken bij het stadium vóór de behandeling), en een subgroepanalyse, waarvan traag vorderende patiënten werden uitgesloten die minder kans maken detecteerbare voordelen te ondervinden van NurOwn. Aangezien deze studie zich in een exploratiestadium bevindt, werd de statistische significantie gedefinieerd als een eenzijdige p-waarde <0,1, met gebruikmaking van de exacte test van Fisher.
ALSFRS-R-responderanalyse, intent-to-treatpopulatie
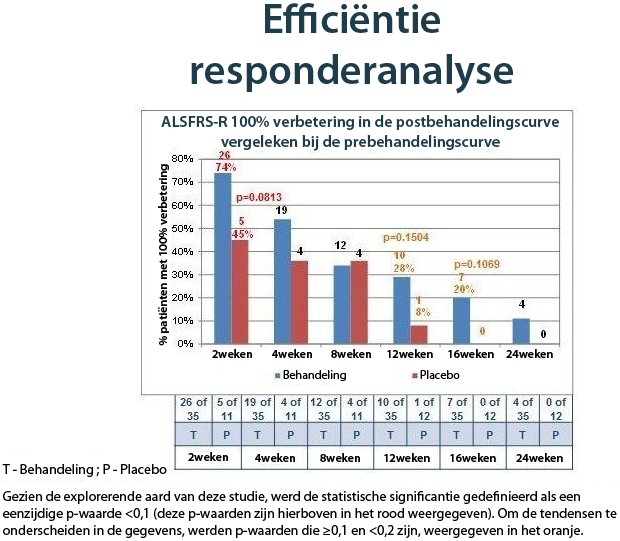
De vooraf bepaalde responderanalyse onderzoekt zowel percentageverbeteringen als absolute puntverbeteringen per maand voor de postbehandelings-ALSFRS-R-curve vergeleken bij de prebehandelingscurve. De eerste spitst zich toe op patiënten die er met 25%, 50%, 75% en 100% op vooruitgingen. Neurologen beschouwen een verbetering met 25% in de curve als enigszins klinisch betekenisvol en een verbetering met 50% in de curve als bijzonder klinisch betekenisvol (Castrillo-Viguera et al, Amyotrophic Lateral Sclerosis 2010; 11: 178_180)2.
Afgezien van alle definities van 'responder' en van alle tijdpunten uitgezonderd één, stelden we een hoger percentage van met NurOwn behandelde deelnemers vast die responders waren vergeleken bij placebo. Bij wijze van voorbeeld: in week 12 (na de behandeling) kende 40% van de deelnemers van de NurOwn-kuur, versus slechts 17% van de placebodeelnemers, een verbetering van de ALSFRS-R-curve na de behandeling van ten minste 50% vergeleken bij de situatie vóór de behandeling.
De grafiek hieronder toont het voordeel dat de NurOwn-behandeling patiënten opbracht. Responders werden gedefinieerd volgens een zeer hoge drempel van 100% verbetering in de postbehandelingscurve vergeleken bij de prebehandelingscurve. Met andere woorden, van een deelnemer werd verwacht dat hij stabiliteit of verbetering vertoonde om als responder te worden beschouwd. De onderstaande grafiek brengt zowel de kortetermijn- als de langetermijnvoordelen in kaart voor met NurOwn behandelde deelnemers in vergelijking met placebo.
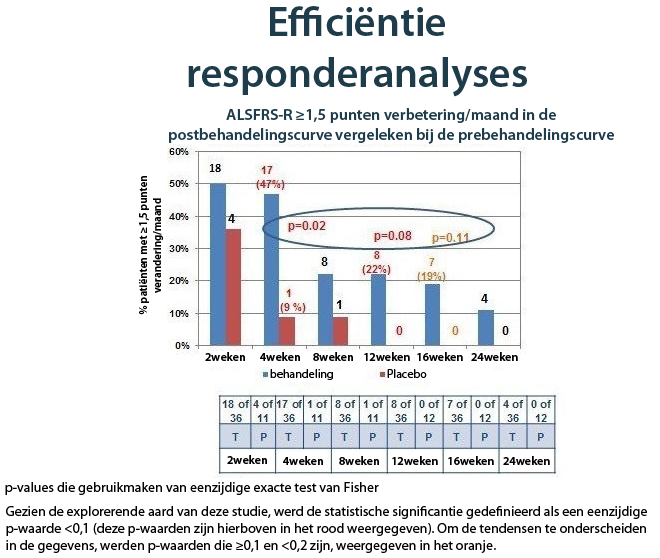
Toen de respons op de behandeling werd geëvalueerd gebaseerd op het absolute punt verbetering/maand in de ALSFRS-R-postbehandelingscurve, vergeleken bij de prebehandelingscurve naarmate de tijd vorderde, vonden de onderzoekers ook hier sterk bewijs van een NurOwn-behandelingseffect. De grafiek hieronder toont de resultaten voor patiënten die een 1,5 of hogere puntverbetering bereikten per maand in de ALSFRS-R-curve gedurende 24 weken, wat een voordeel aantoont ten gunste van NurOwn op elk punt en die statistisch betekenisvol waren na 4, 12 en 16 weken.
ALSFRS-R-responderanalyse, snel vorderende subgroep
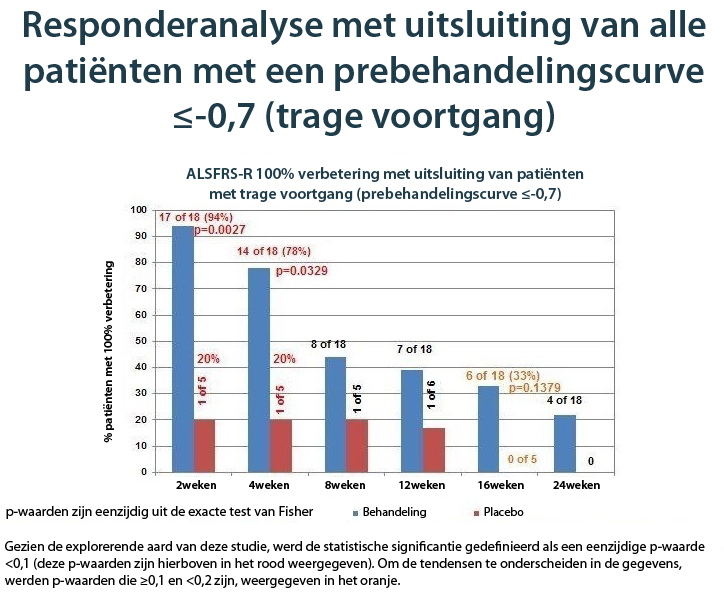
Gezien ervan uitgegaan wordt dat NurOwn de ziektevoortgang vertraagt, werd een vooraf bepaalde subgroep gedefinieerd om deelnemers uit te sluiten bij wie de ziekte traag vorderde (gedefinieerd door een ALSFRS-R-curve van -0,7 of hoger tijdens de prebehandelingsfase). Bij deze deelnemers was de kans kleiner dat NurOwn een meetbaar voordeel vertoonde. De sneller vorderende subgroep omvatte zowat de helft van de deelnemers aan de studie en vertoonde een duidelijk voordeel bij de behandeling met NurOwn: 94% van diegenen die werden behandeld met NurOwn (n=18) bereikten 100% verbetering in de curve na 2 weken, vergeleken bij slechts 20% in de placebo- (n=5) groep (p = 0,0027)(1).Na 4, 8, 12, 16 en 24 weken bedroeg de proportie van responders in de actieve behandelingsgroep vergeleken bij placebo 78% vs. 20%, 44% vs. 20%, 39% vs. 17%, 33% vs. 0% en 22% vs. 0%. De onderstaande grafiek geeft de resultaten weer.
Biomarkeranalyse
Er werden bij de patiënten stalen verzameld van cerebraal-spinaal vocht (cerebral-spinal fluid (CSF)), zoals gespecificeerd in een protocolamendement, nadat de eerste 8 patiënten al waren behandeld. Een totaal van 35 stalen van de pre- en posttransplantatiebezoeken waren beschikbaar voor analyse. De niveaus van neurotrofische en ontstekingsfactoren in elk staal werden gemeten. Een statistisch significante verhoging in de niveaus van VEGF en HGF van pre- tot posttransplantatie werd vastgesteld in de stalen van de patiënten die respondeerden op NurOwn. Er was ook een statistisch significante reductie van ontstekingsmarkers (MCP-1 and SDF-1) gedurende deze periode bij patiënten die werden behandeld met NurOwn, een reductie die niet werd vastgesteld bij de placebogroep.
Veiligheid
NurOwn bleek veilig en goed verdraagbaar te zijn. Het grootste deel van de nadelige gebeurtenissen bleek mild of matig uit te vallen.
Er deden zich geen sterfgevallen voor tijdens de studie en geen enkele patiënt zette de deelname stop wegens een nadelige gebeurtenis. Alle patiënten, zowel die in actieve behandeling als in de placebogroep, hadden last van ten minste één nadelige gebeurtenis die opdook tijdens de behandeling. De nadelige effecten bleken in beide groepen mild tot matig uit te vallen. Nadelige gebeurtenissen die gekoppeld waren aan de behandeling, zoals vastgesteld door de geblindeerde onderzoeker, deden zich iets frequenter voor bij de actief behandelde patiënten dan bij de placebopatiënten, 97,2% vs. 75,0%. De grootste verschillen qua frequentie deden zich voor op het vlak van gelokaliseerde reacties zoals pijn op de plaats van de injectie en rugpijn, en op het vlak van systemische reacties zoals pyrexie, hoofdpijn en arthralgie. Het patroon van nadelige gebeurtenissen is consistent met voorbijgaande reacties op de transplantatie. Deze nadelige gebeurtenissen waren miniem van aard en zelflimiterend. Ernstige nadelige gebeurtenissen (serious adverse events (SAE's)) na de behandeling bleken zich frequenter voor te doen bij patiënten in actieve behandeling (8/36, 22,2%) dan bij placebopatiënten (1/12, 8,3%). De meeste gevallen van SAE waren gekoppeld aan de voortgang van ALS. Meestal ging het daarbij om dysfagie. Geen enkel geval van SAE was gekoppeld aan de studiebehandeling. De risico-voordeelratio voor MSC-NTF-cellen blijft positief.
(1) Alle efficiëntieanalyses gebruikten een eenzijdige alfa=0,10-test
(2) Klinische significantie in de verandering van de achteruitgang in ALSFRS-R, Amyotrophic Lateral Sclerosis. 2010; 11: 178_180. De validiteit van het gebruik van een 'responderanalyse' en het niveau waarvoor eindpuntscores als klinisch betekenisvol bij ALS-patiënten zou worden beschouwd, werd geëxploreerd in een enquête bij 65 clinici gepubliceerd in Amyotrophic Lateral Sclerosis in 2010. "Alle deelnemers aan deze enquête waren het erover eens dat een verandering van 25% of meer in de ALSFRS-R-score op z'n minst enigszins klinisch betekenisvol is. 93 procent van de deelnemers beschouwden een verandering van 50% qua achteruitgang als bijzonder klinisch betekenisvol."
Vertaling: Bart De Becker
Bron: BrainStorm